

¿Quién no se ha enfrentado a lo largo de su vida a montar un puzle? Empezamos desde muy pequeños, con rompecabezas de cubos de pocos elementos que nos permiten construir 6 imágenes distintas con suma facilidad (aunque con pocos años no nos lo parezca). A medida que crecemos nos atrevemos con puzles de 1.000, 5.000, 10.000 piezas, en los que suelen aparecer zonas en las que resulta difícil colocar las piezas porque todas son muy semejantes entre sí (algo típico en un área de cielo o mar). Tanto el número de piezas como estas zonas complejas nos suponen un reto, los puzles simples de la infancia no nos motivan, nos aburren.
En ciencia ocurre todo lo contrario: nos interesa que nuestros rompecabezas, que ahora describen el mundo real, sean lo más sencillos posibles, y si no lo son tratamos de simplificarlos. En este artículo os voy a contar cómo se ha buscado esta simplificación para resolver el gran problema de la secuenciación completa de genomas y para ello voy a utilizar como analogía la resolución de puzles.
Con el abaratamiento de los equipos de nueva generación para la secuenciación completa de ADN, su uso para un diagnóstico o pronóstico de enfermedades está cada vez más cerca de convertirse en cotidiano, pese a las dificultades que aún existen para ello. Recordemos que la secuenciación completa del ADN de un individuo no es más que la lectura de los 3000 millones de pares de bases (cuyos cuatro componentes básicos o nucleótidos suelen representarse por las letras A, G, T y C) que constituyen su genoma. Durante una secuenciación, el genoma a analizar se divide en trozos chiquitines que, tras ser leídos e interpretados, deben unirse entre sí. Esta y otras características hacen que nuestra secuenciación pueda explicarse como el proceso de resolución de un puzle.

Típicamente, a la hora de montar un puzle tenemos una imagen modelo, normalmente en la caja del producto, y pequeñas piezas sueltas que tenemos que colocar para reproducir dicha imagen. Cuando se secuencia el genoma de una persona nos encontramos con una situación similar. Por un lado, tenemos un modelo, el denominado genoma de referencia, que se ha construido a partir de genomas de varios voluntarios y que representa el 99.9% que tenemos en común los seres humanos. Dicho de otra forma, tenemos escrito en un papel (bueno, en un fichero de ordenador) una frase con 3000 millones de letras que sería el modelo para nuestro puzle.

Por otro lado, tenemos el genoma de la persona concreta que nos interesa analizar. Los dispositivos secuenciadores de ADN necesitan romper la cadena de ADN a trocitos pequeños para después poder leer estos (no, no se leen de un tirón los 3000 millones de letras. Al menos aún no). Cuando secuenciamos el genoma de una nueva especie no tenemos ningún modelo previo (ninguna caja con la foto) así que hay que hacer el puzle sin referencia alguna: es lo que se conoce como ensamblado del genoma y es un proceso algorítmico bastante complejo, como ya expliqué en esta charla de Desgranando Ciencia. En el caso de tener un modelo, que es lo que ocurre en el caso de humanos, la situación es semejante a la construcción de un puzle: debemos unir las piezas del ADN de nuestro individuo para que se asemeje al modelo de la caja, es decir, al genoma de referencia.
Pero aquí hay unas cuantas diferencias. En primer lugar, si montamos nuestro puzle de ADN (que ahora veremos que no necesitamos hacerlo) la imagen no será igual que la de la tapa de la caja: somos individuos distintos porque presentamos pequeñas diferencias en nuestro genoma. Pero además, realmente no necesitamos montar nuestro puzle de ADN, ya que únicamente nos interesan las diferencias en nuestro ADN respecto al de referencia: si aparece una mutación, si tenemos un alelo u otro, etc. Así que nos basta con colocar de forma aproximada las piezas en el tablero y quedarnos con aquellas que son diferentes o tienen alguna característica que nos interesa.

Además, hemos de tener en cuenta que se rompen varias cadenas de ADN (y no una sola) que se han de alinear con el mismo genoma de referencia. Es decir, es como si tuviéramos varias cajas con el mismo puzle, hubiéramos mezclado todas las piezas entre sí y tuviéramos que colocarlas sobre el modelo. Para complicar aún más las cosas, los secuenciadores cometen errores al leer algunas letras, así que es como si algunas de nuestras piezas estuvieran manchadas con café.

De esta tarea de colocar los fragmentos de ADN sobre el genoma de referencia (a este proceso le llamamos alineamiento) se encargan programas bioinformáticos denominados alineadores, como por ejemplo bwa o bowtie. Básicamente, estos programas buscan qué zonas del genoma de referencia se parecen más a nuestra pieza de puzle. Un problema reside en que nuestro genoma posee una gran cantidad de zonas con secuencias de letras repetidas, por lo que alinear los fragmentos en estas regiones no resulta nada fácil. Es lo mismo que ocurre con las zonas de mar de nuestro cuadro de Sorolla, en las que las piezas se parecen todas entre sí. Además, el aumento de velocidad de los nuevos secuenciadores trajo consigo una reducción de los fragmentos leídos, lo cual complicó aún más los alineamientos en estas regiones. Esto es semejante a lo que ocurre cuando intentamos colocar en nuestro puzle una pieza de mar: ya es en sí complicado, pero si reducimos el tamaño de la pieza se complica aún más.

Pero hace unos años, la empresa Illumina ideó una solución para resolver este problema. La idea es simple (espero) de entender: se fragmenta el genoma en trozos que se leen desde los dos extremos, sin (necesariamente) llegar a cruzarse las lecturas. Posteriormente, ambas lecturas se alinean con el genoma de referencia, pero teniendo en cuenta que la distancia entre ambas debe coincidir con la longitud del fragmento. Por ejemplo, los fragmentos podrían ser de 500 letras y leerse únicamente 150 letras de cada extremo dejando 200 letras sin leer.
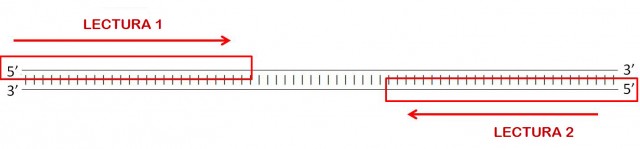
Posteriormente, las lecturas de los extremos de estos fragmentos se alinean con el genoma de referencia. Pero fijémonos en que no basta con que coincidan con el modelo estas 150 letras de cada extremo, sino que además la distancia entre ellos ha de ser de 200 letras (salvo inserciones o borrados).
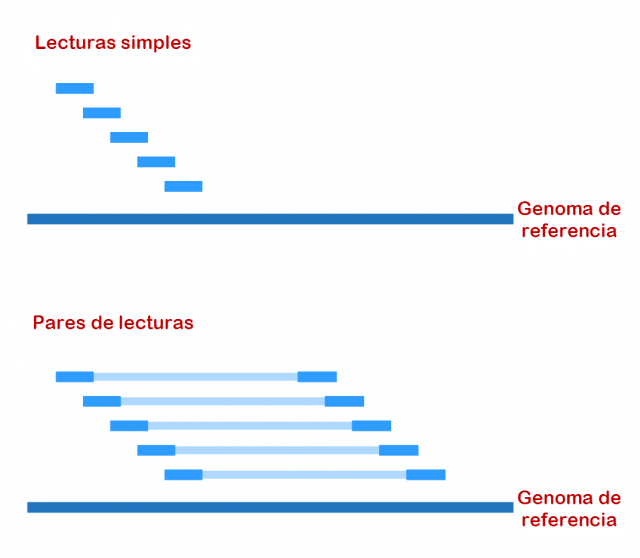
Para entenderlo mejor, vamos a ver cómo sería la equivalencia con nuestro puzle de Sorolla. Sería como si tuviéramos pares de piezas unidos por un segmento rígido con la distancia real en la solución del puzle. De esta forma, sería más sencillo colocar la pieza del mar si la otra pieza a la que estuviera conectada fuera más simple de localizar.

Esta tecnología de Illumina, conocida como paired-end reads (lecturas de parejas de extremos) ha contribuido a obtener genomas secuenciados de alta calidad y más fáciles de alinear con el genoma de referencia.
Pero la solución a nuestro problema de montar el rompecabezas en la región del cielo tiene otra solución. ¿Y si las piezas que fabricáramos fueran mucho más grandes? En ese caso sería muchísimo más sencillo montar nuestro puzle. De hecho, y si fueran suficientemente grandes, ni siquiera necesitaríamos el modelo.

Esto es lo que ocurre con los nuevos secuenciadores de fragmentos largos, como los de PacBio o Oxford Nanopore. En este caso, los fragmentos leídos pueden llegar a ser de varias decenas de miles de letras, con lo que el genoma prácticamente sale ensamblado sin comparar con el genoma de referencia.
Aunque pueda parecer que el problema de obtener alineamientos de calidad en regiones muy repetitivas del genoma estaría resuelto con esta tecnología, lo cierto es que aún es muy cara y solo accesible a grandes centros de investigación, por lo que los secuenciadores de Illumina y sus lecturas por parejas siguen siendo los de uso más habitual.
Más información
A continuación se incluyen dos vídeos en los que se muestra con algo más de detalle cómo funcionan las tecnologías de secuenciación de Illumina y PacBio.
Soy doctor en ciencias químicas, e inicié mi investigación y doctorado en el campo de la química cuántica. Actualmente soy profesor titular de informática en la Universitat Jaume I de Castellón y colaboro como bioinformático con el grupo «Biología de retrotransposones» del centro de genómica y oncología GENYO de Granada. Mi investigación se centra en el estudio de los elementos genéticos móviles y microARN, así como su influencia en tumores y en enfermedades concretas como el síndrome de deleción 22q11.