

En un artículo anterior de Naukas os hablé del genoma de referencia humano y cómo su uso ha sido fundamental para los avances en genética y medicina personalizada de las últimas dos décadas. El genoma de referencia actúa como modelo a la hora de unir los millones de pequeñas piezas que se obtienen tras la secuenciación de un genoma individual y nos permite encontrar diferencias que apunten a cambios genéticos relevantes para una patología concreta. La última versión del genoma de referencia, de febrero de 2019, se conoce como GRCh38.p13 y puede descargarse aquí. Pero, como ya se comentó en el artículo anterior, el genoma humano de referencia presenta un par de problemas importantes.
En primer lugar, contiene huecos, vacíos de los que no conocemos su composición en áreas difíciles de secuenciar (normalmente porque en ellas hay un elevado número de repeticiones). En la última versión del genoma de referencia (GRCh38.p13) el número de huecos asciende a 349, lo cual no está nada mal si pensamos que en el primer borrador publicado en 2001 había cerca de 150 000. Este problema está siendo abordado por el T2T consortium, que ya ha publicado una versión completa del cromosoma X y tiene pendiente de revisión por pares la publicación del cromosoma 8.
El otro problema es que el actual genoma humano de referencia, incluso tras rellenar los huecos existentes, tampoco sería una representación fidedigna del genoma de la mayoría de grupos poblacionales, ya que se construyó a partir de muestras de ADN de unos pocos individuos. Como ya se comentó en el artículo anterior, cerca del 70 % de la referencia se basa en un solo individuo de origen caucásico-africano. Esto hace que nuestro ADN difiera en gran medida del genoma de referencia; incluso se han encontrado personas que tendrían secuencias nuevas (ausentes en la referencia) que sumarían hasta 14 200 000 bases (14.2 Mb). Algunos estudios sugieren que las secuencias de ADN humano no incluidas en el genoma de referencia estarían en un amplio rango en torno a los 100 Mb. Es más, incluso en el caso de secuencias que sí aparecen en una gran parte de la población, en ocasiones la variante registrada en el genoma de referencia no sería la más común; por ejemplo, si en una determinada posición del genoma la «letra» más habitual en la población es la C, en el genoma de referencia podría haber una T.
¿Cómo solucionar este problema? Uno de los esfuerzos actuales de la comunidad científica se orienta a la construcción de un pangenoma, un genoma que nos represente a todos.
El pangenoma
El genoma de cualquiera de nosotros podría representarse como una larga secuencia de unas 3 200 000 000 letras, que se leería de izquierda a derecha como un libro (me gusta la comparación de que un genoma humano impreso ocuparía lo mismo que 1 500 libros de El Quijote). El genoma de referencia humano actual se representa de la misma forma. Por ejemplo, aquí tenéis un pequeño fragmento del cromosoma X:

Para empezar a construir un pequeño pangenoma casero, nosotros utilizaremos una secuencia corta de 17 nucleótidos:

Imaginemos que este es nuestro genoma de referencia actual, al que queremos ir añadiendo las variaciones que vayamos encontrando en los genomas de nuevos individuos. Como vamos a utilizar grafos para construir este pangenoma (¡no huyáis, bellacos!) un primer paso es representar esta primera secuencia como un grafo sencillo (podría utilizar flechas para unir estos nodos, ya que la dirección de lectura es de izquierda a derecha, pero no lo haré para no complicar la explicación):

A partir de esta representación inicial podemos añadir nuevas variantes como caminos en este grafo. Por ejemplo, imaginemos que encontramos en la población una nueva secuencia en la que encontramos en la séptima posición una G en lugar de la C de la primera secuencia de referencia (marcada en la siguiente figura con un color distinto). En ese caso podemos añadir un nuevo «camino» que represente a esta nueva secuencia.

Podría ocurrir también que la diferencia con la secuencia inicial fueran varios nucleótidos añadidos en una posición concreta. En la siguiente figura se muestra una secuencia de este tipo (con los nuevos nucleótidos respecto a la referencia en un color distinto) y cómo los añadiríamos a nuestro grafo creando un nuevo camino. A este proceso por el que aparecen nuevos nucleótidos en una posición concreta le llamamos inserción.
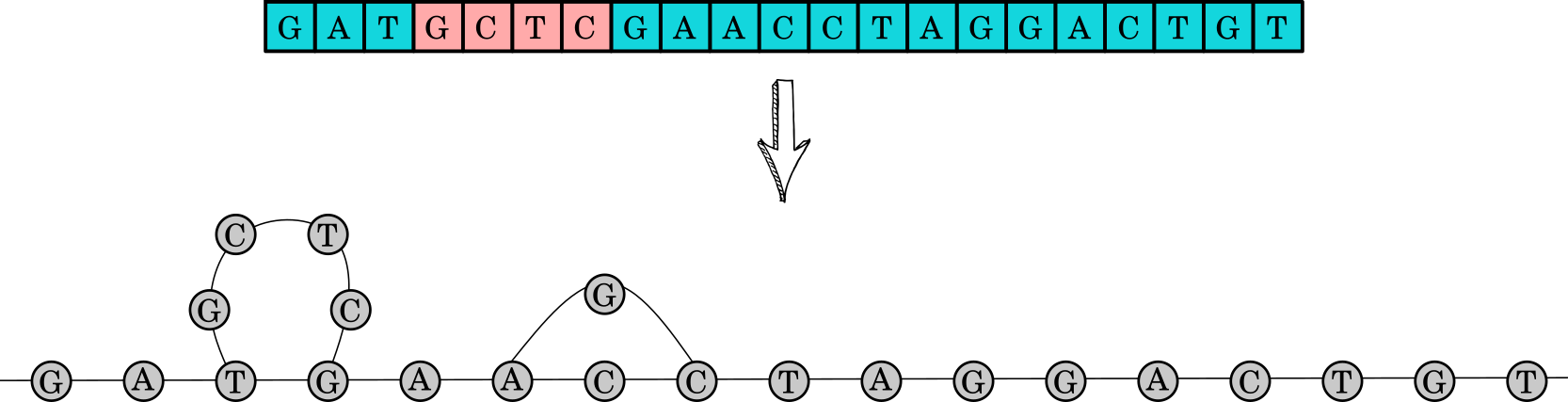
Por último, también podría ocurrir que encontráramos una secuencia en la que faltaran nucléotidos de la secuencia de referencia, como la que se muestra en la siguiente imagen (se han añadido unos cuadros vacíos para indicar que faltan letras respecto a la referencia). Para representar este proceso de borrado podríamos incluir un nuevo camino que «saltara» los nucleótidos ausentes.

Con tantos caminos, ocurrirá con mucha frecuencia que nuestro grafo acabe incluyendo secuencias que realmente nunca hemos encontrado. Por ejemplo, el camino que se señala en rojo en la siguiente imagen no se corresponde con ninguna secuencia descrita hasta el momento.

Para evitar que se recorran caminos que no se corresponden con secuencias reales podemos utilizar colores para los nuevos nodos que vayamos añadiendo, de forma que sólo podremos recorrer caminos en los que los nodos coloreados tengan el mismo color.
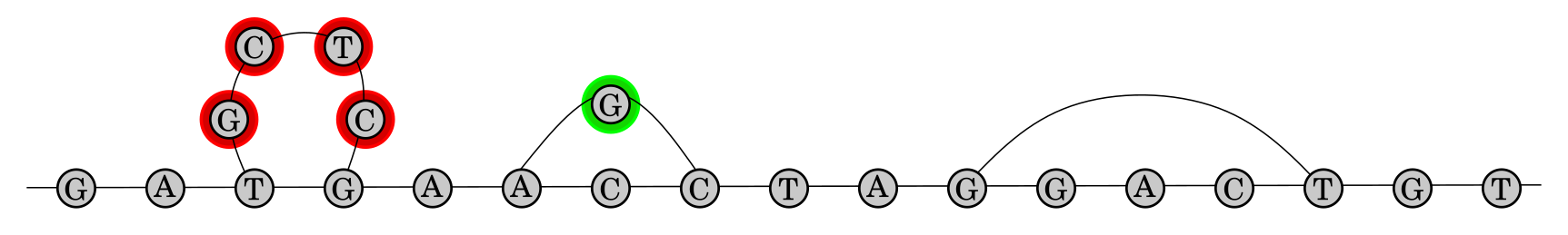
De esta forma, si estamos comparando con nuestra referencia una nueva secuencia y se asemeja al fragmento con nodos rojos, ya no seguiríamos el camino con el nodo verde. De esta forma, y si usáramos secuencias de distintos grupos poblacionales, podríamos obtener un pangenoma que sería más representativo.
Existen herramientas que nos permiten representar pangenomas de forma más «agradable». La representación que se muestra a continuación, por ejemplo, se ha obtenido con la aplicación sequenceTubeMap, que muestra el pangenoma imitando a un plano de metro, de forma que siguiendo cada línea por su color recuperamos la secuencia original. Por ejemplo, la secuencia de ADN con la que se construyó la línea naranja es:
TTAACCATGAACGAGGTG

Pese a que este tipo de representaciones de pangenomas en forma de grafos se están utilizando con éxito, no por ello están exentas de problemas. Un primer problema es que el almacenamiento en memoria de estos pangenomas ocuparía más; no sólo porque contendría muchos más nodos que el genoma lineal, sino también porque cada nodo tendría que reservar más memoria para un «color». Además, aunque no he hablado de ello por no complicar la explicación, sería conveniente que cada nodo almacenara además un valor con la frecuencia con la que se encuentra en la población dicha variante.
Otro problema no menos importante es que prácticamente la totalidad de software que tenemos para comparar secuencias de ADN con el genoma de referencia (lo que se conocen como alineadores) están basados en secuencias lineales y no en grafos. Todo este software ha sido comprobado y analizado en cientos de investigaciones, por lo que el paso a un software nuevo no sería inmediato.
No obstante, el futuro de los genomas de referencia pasa indudablemente por su representación en forma de pangenoma. Para terminar, os aconsejo este vídeo en el que se explican algunos de los conceptos explicados en el presente artículo (en inglés).
Referencias
- Sherman, R.M., Salzberg, S.L. Pan-genomics in the human genome era. Nat Rev Genet 21, 243–254 (2020). https://doi.org/10.1038/s41576-020-0210-7
- Golicz, Agnieszka A., et al. Pangenomics comes of age: From bacteria to plant and animal applications. Trends in Genetics 36.2 (2020): 132-145. https://doi.org/10.1016/j.tig.2019.11.006
- Jordan M. Eizenga, J.M. et al. Pangenome graphs. Annual Review of Genomics and Human Genetics 2020 21:1, 139-162. https://doi.org/10.1146/annurev-genom-120219-080406
Soy doctor en ciencias químicas, e inicié mi investigación y doctorado en el campo de la química cuántica. Actualmente soy profesor titular de informática en la Universitat Jaume I de Castellón y colaboro como bioinformático con el grupo «Biología de retrotransposones» del centro de genómica y oncología GENYO de Granada. Mi investigación se centra en el estudio de los elementos genéticos móviles y microARN, así como su influencia en tumores y en enfermedades concretas como el síndrome de deleción 22q11.