

Suele ocurrir en la vida diaria que tenemos problemas para darnos cuenta de las cosas importantes. Esta experiencia ha sido común en tiempos de confinamiento, pero es habitual en todos los ámbitos de la vida, no ver lo que está detrás de lo evidente. Este es el caso de una molécula, la coenzima Q o…
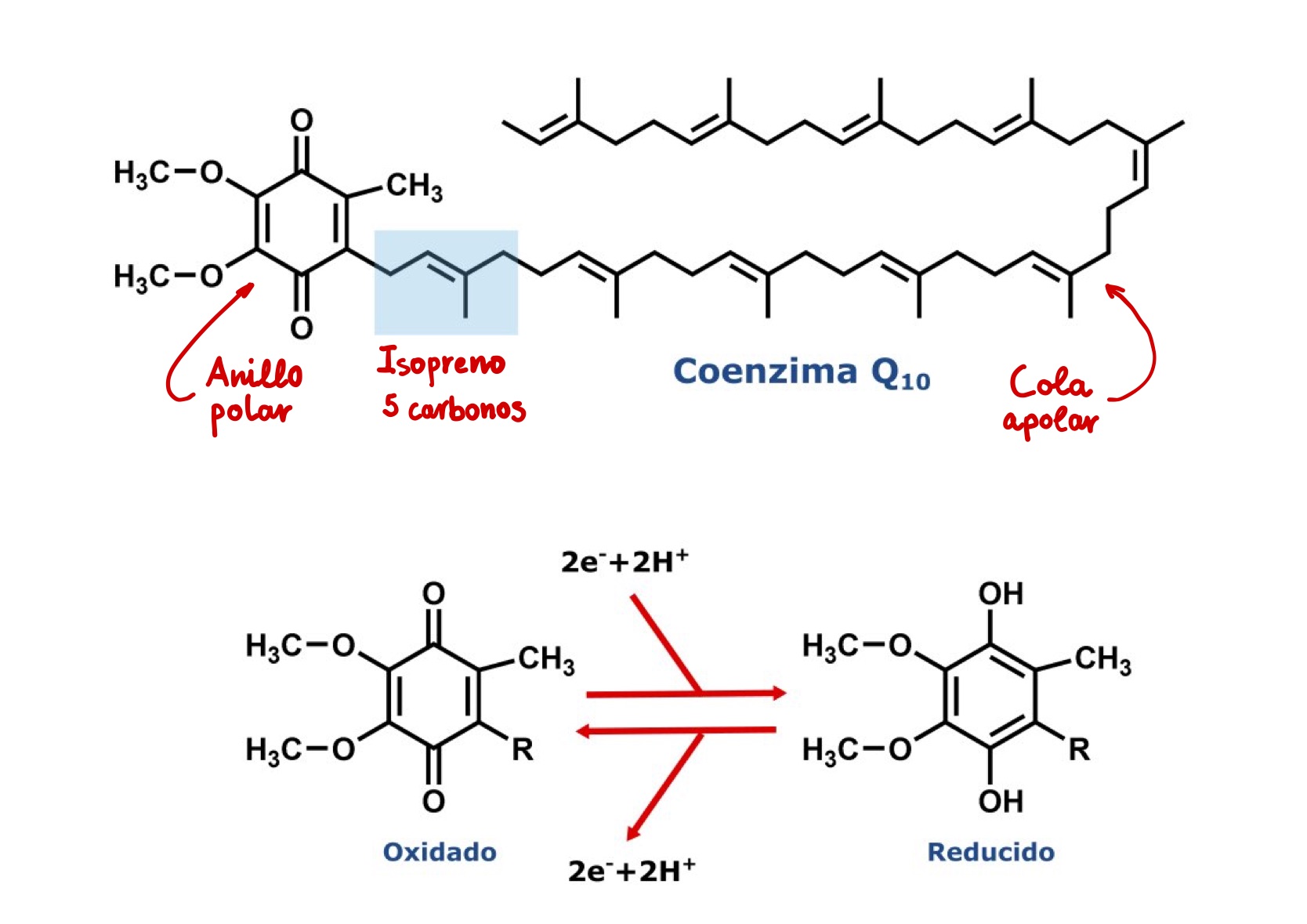
Figura 1. Estructura y función en la coenzima Q10. La coenzima Q10 es un lípido muy especial. Se compone de una cola isoprenoide apolar de 50 carbonos, en la especie humana. Estos carbonos se agrupan en unidades de 5 carbonos denominadas isopreno (sombreado en azul). Por tanto, los humanos sintetizamos (sí, usted) una cola de 10 isoprenos, CoQ10. En otras especies el tamaño es diferente, entre 6 y 10 isoprenos, pero no existe ninguna relación entre el tamaño de la cola isoprenoide y la posición evolutiva de la especie. La longitud de la cola isoprenoide está relacionado con la estructura de la membrana mitocondrial y las proteínas que allí se localizan. La función de la cola isoprenoide es insertar y mantener al CoQ10 en el interior de la membrana mitocondrial. El segundo componente es el anillo polar, un anillo aromático con dos grupos cetona dispuestos de forma enfrentada (quinona) y sustituido con grupos metilo y metoxi. Los grupos cetónicos del CoQ10 pueden aceptar y donar electrones, lo que explica las funciones del CoQ10 como transportador de electrones y como antioxidante en las membranas celulares. La forma oxidada o ubiquinona se convierte en la forma reducida o ubiquinol cuando acepta 2 electrones y 2 protones. Este este mecanismo es reversible, y se utiliza cuando el CoQ10 funciona en la cadena de transporte de electrones de la mitocondria. Como antioxidante en las membranas puede regenerar a otros antioxidantes (vitamina E o vitamina C), o destruir directamente a los radicales libres.
Suele ocurrir en la vida diaria que tenemos problemas para darnos cuenta de las cosas importantes. Esta experiencia ha sido común en tiempos de confinamiento, pero es habitual en todos los ámbitos de la vida, no ver lo que está detrás de lo evidente. Este es el caso de una molécula, la coenzima Q o ubiquinona, que tiene una historia paquidérmica más allá de su presencia en cosméticos o suplementos vitamínicos.
Cualquier descubrimiento importante tiene siempre una curiosa historia detrás, y la coenzima Q o ubiquinona la tienen. Y está relacionada con este empeño mío de repetir ambos nombres. Ya se sabe, ¿a quién quieres más? ¿A papá o a mamá? En el año 1955 el grupo de Dr. Morton en Liverpool aisló una sustancia de naturaleza lipídica de células intestinales de caballo (1), que al ser insaponificable debía pertenecer a la familia del colesterol o mejor a la familia de los isoprenoides.
Lo más característico era que esta sustancia tenía un pico de absorción a 272 nm, y además comprobaron que se podía aislar de distintos tipos de organismos, incluso no relacionados filogenéticamente. Como tenía propiedades de quinona (un anillo aromático de benceno con dos grupos cetónicos opuestos) y estaba en todas partes, adoptaron el nombre de ubiquinona, quinona que está en todos lados. Como ahora veremos, estos descubrimientos llegaron tras la aparición de dos nuevos equipos en los laboratorios bioquímicos, la ultracentrífuga de gran capacidad, necesaria para purificar las mitocondrias y otros componentes subcelulares, y el espectrofotómetro de doble haz, fundamental para determinar de forma precisa los picos de absorción y los cambios en las moléculas generado por su estado de oxidorreducción. Dos años después, en el laboratorio del Dr. Green en la Universidad de Wisconsin, Frederick Crane trabajando con mitocondria de coliflor y de músculo cardiaco de ternera aisló una molécula que denominó Q275, al mostrar un pico de absorción a esa longitud de onda (2).
Esta molécula, que tenía propiedades de quinona, se encontraba asociada con gran afinidad a las proteínas mitocondriales y por ello tuvo que extraerse con solventes orgánicos. Como hecho novedoso, la capacidad de la molécula para oxidarse y reducirse en condiciones fisiológicas permitió deducir que participaba en el transporte de electrones en la mitocondria (Figura 1). Todo esto sugirió un nombre para la molécula, la coenzima Q, otro nombre para la ubiquinona. El concepto coenzima proviene de su función en el consumo de oxígeno mitocondrial, la retirada de la coenzima Q de la mitocondria eliminaba el consumo de oxígeno, que se restauraba con la adición de la coenzima Q.
Es verdad que el concepto Q proviene del término quinona pero también de la moda de denominar a las vitaminas con una letra. En este caso, Q. Ya en 1958, la colaboración con el Dr. Folkers en los laboratorios de investigación de la compañía Merck en Rahway (New Jersey) permitió completar la estructura del coenzima Q10 (el coenzima Q humano) y su compleja síntesis (3, 4). ¿Pero quién la descubrió realmente? Depende, pero en todo caso siempre ha estado ahí ya que no sólo es una molécula ubicua, también es antigua, y evolutivamente insustituible.
A pesar de lo anterior, la gloria para el coenzima Q (aquí podéis descubrir cuál es mi opinión sobre su descubridor) llegó algo después, cuando Peter Mitchel formuló el mecanismo quimiosmótico que explicaba la síntesis de ATP en la mitocondria, y donde el coenzima Q tenía reservado un papel estelar (5), primero en la propia denominación en inglés, protonmotive Q-cycle (6) y segundo, por la obtención por parte de Mitchel del Premio Nobel en 1978. La importancia de la coenzima Q y el mecanismo quimiosmótico es la síntesis de ATP. En estos momentos de cambio climático asistimos al cambio del paradigma energético, a la migración desde fuentes no renovables a las renovables.
Sin embargo, tenemos un problema que la naturaleza ya solucionó hace millones de años cuando apareció el oxígeno en la Tierra. La producción de energía renovable en este momento está desacoplada, no se puede sincronizar la producción de energía con el consumo. Tan sólo un tipo de energía renovable, la hidroeléctrica se puede acoplar. Recuerden este detalle para más delante. Las células tienen un truco. Con la energía producida por la combustión controlada de los alimentos (carbohidratos, grasas y en algunos casos, proteínas) las células fabrican ATP, una molécula estable que se puede transportar a cualquier sitio en la célula para ser usada de forma asíncrona a su producción. Esta fabricación de ATP se produce mayoritariamente en la mitocondria de las células, y para ello necesitamos a la coenzima Q.
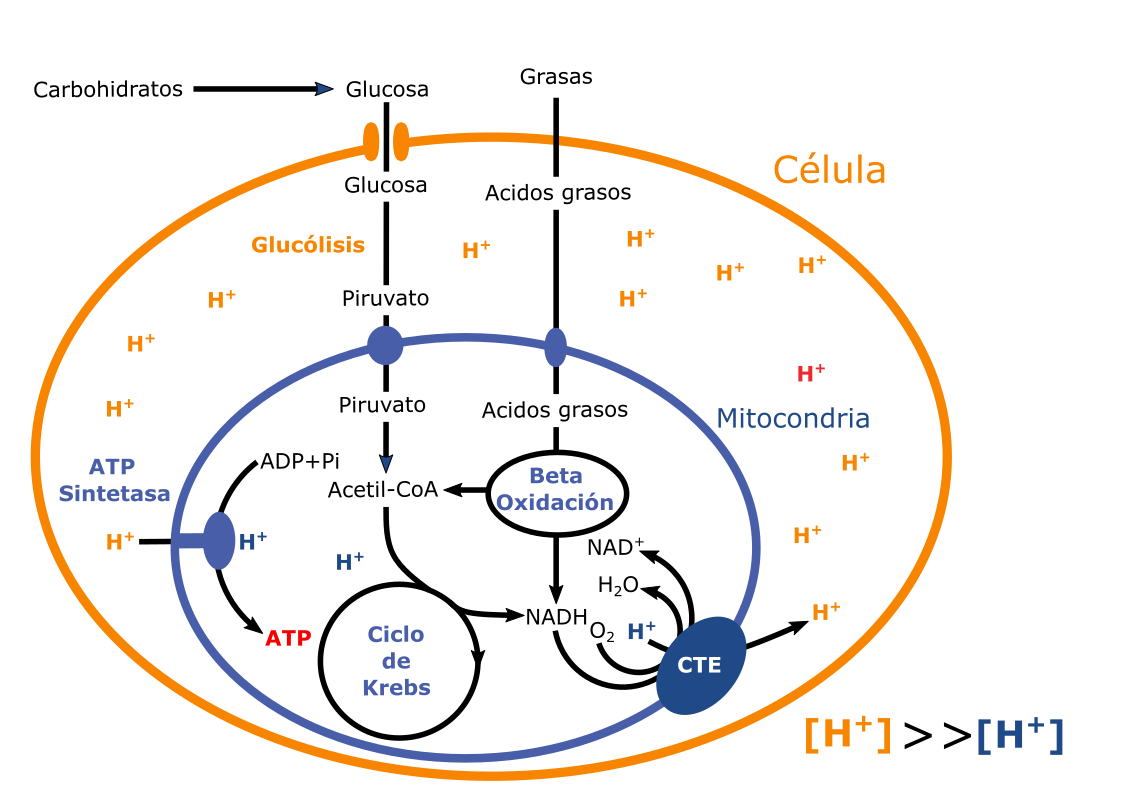
Figura 2. La teoría quimiosmótica y la producción de ATP la mitocondria. En el esquema se muestra un resumen de las rutas de producción de energía de la célula. Se trata de un esquema simplificado y generalista. Simplificado al mostrar las rutas más importantes y generalista ya que se refiere a una célula ideal. Los nutrientes con función energética (carbohidratos y grasas), se introducen en la célula mediante el uso de transportadores. Los carbohidratos, representados por la glucosa, sufren una degradación parcial hasta piruvato en el citoplasma, pero la auténtica degradación tiene lugar en la mitocondria. Tanto el piruvato como los ácidos grasos que provienen de las grasas son convertidos en acetil-CoA en la mitocondria. El primero mediante la piruvato deshidrogenasa y los segundos mediante la beta-oxidación. Este acetil-CoA se utiliza en el ciclo de Krebs para extraer todo el poder reductor posible en forma de electrones, que se acumulan como NADH. El NADH es una forma de almacenar la energía de los electrones provenientes de los alimentos. La segunda fase del metabolismo bioenergético mitocondrial se inicia en la cadena de transporte electrónico (CTE) donde el flujo de electrones entre sus componentes (simplificado en el esquema) libera energía libre que se usa para bombear protones en contra de gradiente (subir agua a la presa). Ello permite la formación de un gradiente de protones, una mayor concentración de protones fuera de la mitocondria que dentro. Esto prepara el camino de la tercera fase, la entrada de los protones a la mitocondria a favor de gradiente (salida del agua de la presa) con la ayuda de la ATP-sintetasa, que usa la energía liberada para producir ATP en el interior de la mitocondria).
En la mitocondria se producen reacciones químicas que degradan los nutrientes presentes en los alimentos (Figura 2). Los carbohidratos son degradados parcialmente fuera de la mitocondria por la glucólisis, pero los restos de esta reacción se transportan a la mitocondria para ser convertidos en energía mediante el ciclo de Krebs, ciclo del ácido cítrico o ciclo de los ácidos tricarboxílicos. Cualquier nombre es bueno. Un proceso similar es sufrido por las grasas, que son degradados en la mitocondria mediante un proceso denominado beta-oxidación. Estas reacciones son reacciones de oxidación, donde los nutrientes pierden electrones.
Un electrón es una partícula subatómica que por una parte contiene mucha energía, y por otra nunca está libre en la célula. Sería como liberar una piraña en un acuario de peces payaso. Por ello, los electrones se acumulan en moléculas como el NADH, los sumideros celulares del poder reductor, un poder muy energético. La mitocondria posee un sistema para convertir los electrones acumulados en el NADH en energía en forma de ATP, y se denomina cadena de transporte de electrones (CTE) o cadena respiratoria mitocondrial, unas proteínas localizadas en la membrana interna de la mitocondria. En esta cadena los componentes aceptan electrones de uno de ellos y lo ceden al siguiente como la cadena de cubos de agua para apagar un fuego. El NADH es la molécula que cede los primeros electrones y el oxígeno es el aceptor final en la cadena, que se convierte en agua. Lo más crítico es que en cada paso el eslabón de la cadena que cede el electrón tiene más energía que el aceptor, y esta diferencia de energía se libera al sistema, pero se utiliza para producir un trabajo útil, el transporte de un protón (H+) desde el interior de la mitocondria (matriz) al exterior de ésta. La clave de este sistema es la impermeabilidad de la membrana mitocondrial a los protones, como no pueden atravesar la membrana libremente para volver a la matriz se acumulan en el exterior de la mitocondria y generan un gradiente electroquímico. Químico al elevar la concentración de protones fuera de la mitocondria, y eléctrico ya que al mostrar carga positiva se genera un espacio exterior más positivo que la matriz mitocondrial. La existencia de este gradiente es la base de la teoría quimiosmótica.
Pero ¿dónde está aquí el ATP? Bueno, los protones pueden volver a la matriz a través de un prodigio de máquina molecular, la ATP sintetasa. Cuando los protones se introducen en la matriz lo hacen a favor del gradiente electroquímico (por carga al ir a un sitio negativo y por concentración al ir donde hay menos protones), y ello libera energía que usa la ATP sintetasa para producir ATP desde ADP y fosfato libre. Recuerden el ejemplo que indiqué aguas arriba, la presa hidroeléctrica. Acumulamos agua en la presa (un gradiente) con mucha energía potencial que se libera cuando se abre la compuerta. Esta energía se usa para impulsar alternadores que generan electricidad. Energía potencial convertida en eléctrica, o energía potencial convertida en energía química en la mitocondria. Todo esto también generó otro Nobel, en 1997 para Paul Boyer en UCLA.

Figura 3. La función clave de la coenzima Q10. La estructura de la coenzima Q10 le permite introducirse entre las dos capas de fosfolípidos de la membrana mitocondrial interna. La cola isoprenoide estabiliza la molécula mientras permite a la cabeza polar cierta movilidad, que depende de su estado redox. En la CTE de la mitocondria, el NADH cede sus 2 electrones al complejo I. Este complejo es una estructura transmembrana compuesta por 45 subunidades que canaliza los electrones hacia una molécula de CoQ10 en estado oxidado (rojo), que al reducirse (negro) modifica su orientación en la membrana desde la matriz hacia el exterior. Las moléculas reducidas se mueven en la membrana para interaccionar con otros componentes de la CTE. En este ejemplo, con el complejo III. La coenzima Q10 reducido (negro) cede los electrones al complejo III y se oxida (rojo) para cerrar el ciclo de transporte. El complejo III usa los electrones para reducir al citocromo c, un segundo transportador de electrones de la CTE, pero de tipo soluble. El coenzima Q10 muestra aquí su importante función, transportar electrones a través de un membrana que es impermeable a las moléculas o partículas cargadas, como es el caso de los electrones y los protones.
Y ahora es el turno de la coenzima Q. En la cadena de transporte mitocondrial los electrones deben ser transportados desde la cara interna de la membrana mitocondrial y ser cedidos a componentes que está localizados en la cara externa de la mitocondria. Deben cruzar la membrana mitocondrial, una membrana compuesta por lípidos que no dejan pasar a partículas cargadas como los electrones (Figura 3). Esa es la función de la coenzima Q, transportar los electrones en el entorno lipídico y apolar de la membrana. Permitir que los protones puedan ser transportados al exterior, generar el gradiente electroquímico, y permitir la acción de la ATP sintetasa. Nada más y nada menos.
Y esto es el elefante, generar ATP es una función enormemente importante para nuestra vida. Mutaciones en los genes que codifican las proteínas encargadas de sintetizar la coenzima Q causan el síndrome de deficiencia de CoQ10, una enfermedad rara mitocondrial de efectos devastadores, y que conduce a la muerte del individuo cuando el nivel de síntesis es nulo o muy reducido. Afortunadamente los casos de muerte por deficiencia de coenzima Q no son muy numerosos, pero existen otras funciones de la coenzima Q que se ponen de manifiesto cuando la deficiencia es menos acusada. Las otras funciones afectadas no son tan generales y sistémicas, pero sí son muy numerosas e incrementan la complejidad del diagnóstico y del tratamiento. Estos problemas se colocan bajo la sombra del elefante, y requieren contar otra historia sobre la coenzima Q. A pesar de todo hay esperanza, la deficiencia de coenzima Q es la única enfermedad mitocondrial que se puede tratar mediante suplementación. Ya saben de qué. Toda una esperanza.
Este artículo nos lo envía Carlos Santos Ocaña «Soy profesor titular de Biología Celular de la Universidad Pablo de Olavide. Imparto docencia en Biotecnología, Nutrición Humana y Dietética, y en el Máster de Biotecnología Sanitaria. Mi investigación se ha centrado siempre en el estudio de las funciones de la coenzima Q, y en los mecanismos de regulación de su síntesis. Actualmente investigo en el diagnóstico, mecanismos moleculares y terapia de las enfermedades raras mitocondriales generadas por una deficiencia de coenzima Q10. Soy investigador adscrito al CIBER de Enfermedades Raras, un centro de investigación en red dependiente del Instituto de Salud Carlos III, y mi laboratorio está emplazado en el Centro Andaluz de Biología del Desarrollo- CABD, un instituto mixto del CSIC, Junta de Andalucía y Universidad Pablo de Olavide, en Sevilla. Podéis seguir a Carlos en twitter en @CarlosSantosOc y también en el Grupo @BioCelUPO investigando enfermedades raras.
Referencias científicas y más información:
- FESTENSTEIN, G. N., HEATON, F. W., LOWE, J. S., and MORTON, R. A. (1955) A constituent of the unsaponifiable portion of animal tissue lipids (lambda max. 272 m mu). Biochem. J. 10.1042/bj0590558
- Crane, F. L., Hatefi, Y., Lester, R. L., and Widmer, C. (1957) Isolation of a quinone from beef heart mitochondria. BBA – Biochim. Biophys. Acta. 25, 220–221
- Wolf, D. E., Hoffman, C. H., Trenner, N. R., Arison, B. H., Shunk, C. H., Linn, B. O., McPherson, J. F. J. F., and Folkers, K. (1958) Coenzyme Q. I. Structure studies on the coenzyme Q group. J. Am. Chem. Soc. 80, 4752
- Shunk, C. H., Linn, B. O., Wong, E. L., Wittreich, P. E., Robinson, F. M., and Folkers, K. (1958) COENZYME Q. II. SYNTHESIS OF 6-FARNESYL- AND 6-PHYTYL-DERIVATIVES OF 2,3-DIMETHOXY-5-METHYLBENZOQUINONE AND RELATED ANALOGS. J. Am. Chem. Soc. 80, 4753–4753
- MITCHELL, P. (1961) Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature. 191, 144–148
- Mitchell, P. (1975) The protonmotive Q cycle: A general formulation. FEBS Lett. 59, 137–139





